 WUHAN University Website
WUHAN University WebsiteCatalytic Asymmetric Diels-Alder/Formal [4+2]-Cycloaddition
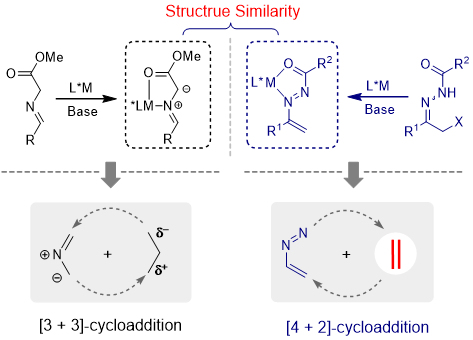
Azoalkene can be readily in situ-generated from the a-halogeno hydrazone, which have been commonly employed as key intermediates for the synthesis of various five-membered achiral dinitrogen-containing heterocycles. Comparing the structure similarity between metallo-azomethine ylide and metallo-azoalkene, we believed that six-membered heterocycles bearing biologically interesting tetrahydropyridazine moiety could be constructed through formal [4+2] reaction of azoalkene with some electron-rich compounds.
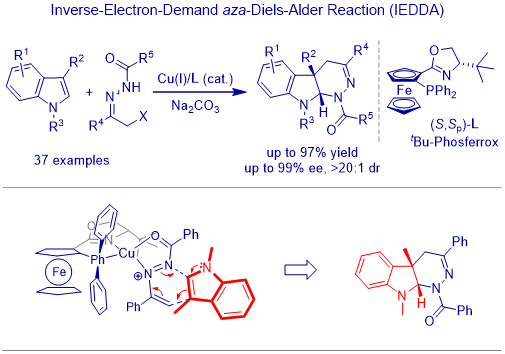
In classical organic chemistry, indole is classfied as a p-excessive heterocycle and it nucleophilicity on C3-position has been well studied both in achiral and chiral reaction. Along with the dvelopment of enamine/iminium chemistry, indole derivatives have received much attentions in the study of cascade reactions. Catalytic asymmetric C2,C3-annulation of indoles, which involves both the nucleophilicity on C3 (enamine-type reactivity) and electrophilicity on C2 (iminium-ion-type reactivity), has become one of the most powerful and diversity-oriented syntheses for constructing enantiomerically enriched [2,3]-fused indoline derivatives. We have successfully developed Cu(I)-catalyzed asymmetric inverse-electron-demand aza-Diels-Alder reaction of indoles with in situ-formed azoalkenes for the direct construction of biologically attractive and enantiopure [2,3]-fused indoline heterocycles.
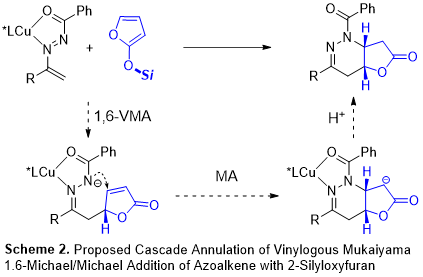
Elaboration of 2-silyloxy furans as the readily-accessible nucleophilic synthons of the g-anion of 2(5H)-furanone by means of vinylogous Mukaiyama-Aldol, Mukaiyama-Michael, and Mukaiyama-Mannich type additions has been the well-established method via electrophilic attack at C-5 position. In consideration that electron-deficient unsaturated lactone moiety in butenolide is a potential Michael acceptor easily trapped by an inbuilt nucleophile group, it is believed that 2-silyloxy furans could be utilized as dipole-type synthons in cascade reaction by sequentially reacting as a nucleophile and an electrophile, giving rise to fused butyrolacton (Scheme 1). This cascade approach involves the nucleophilicity on C5 of the 2-silyloxy furan and the electrophilicity of C4 of the formed butenolide. Surprisingly, however, this kind of asymmetric cascade annulation with 2-silyloxy furans has received much less attention despite the numerous examples of butyrolactone stereogenicity found in natural alkaloids and biologically active compounds.

We envisaged that the combination of 2-silyloxy furans and azoalkenes could constitute an unprecedented formal [4 + 2] cycloaddition through a cascade protocol. As shown in Scheme 2, the initial vinylogous Mukaiyama 1,6-Michael addition of 2-silyloxy furan to the in-situ formed metallo-azoalkene affords an butenolide intermediate. Subsequently, the negative charged N would attack the electron-deficient unsaturated lactone via Michael addition followed by protonation to afford the fused butyrolactons.
We have successfully developed an unprecedented Cu(II)-catalyzed asymmetric cascade Mukaiyama 1,6-Michael/Michael addition of 2-silyloxy furans with in situ-formed azoalkenes. The key feature of the current methodology is that furan-based dienoxy silanes could be utilized as efficient dipole-type synthons. This cascade annulation process provides a straightforward approach to a variety of biologically important and structurally complicated fused-butyrolactones in good yield with high regioselectivity and excellent stereoselectivity. The studies of carbon isotope effects measured by 13C NMR indicated a stepwise mechanism for this annulation.

While significant progresses have been achieved with indoles as aromatic dienophiles, furans have received much less attention in such kind of synthetic transformation. In general, furans have been widely employed as dienes in normal electron-demand [4+2] Diels-Alder reactions with alkenes, alkynes or allenes, [4+3] annulation with 2-oxyallyl cations or vinylcarbenoids. The dienophilic behavior of furans was sporadically addressed only in non-asymmetric transformations. Therefore, developing asymmetric reaction with furans as dienophiles is challenging probably due to the strongly enophilic nature and multiselectivity issues such as chem-, regio- and stereoselectivity.
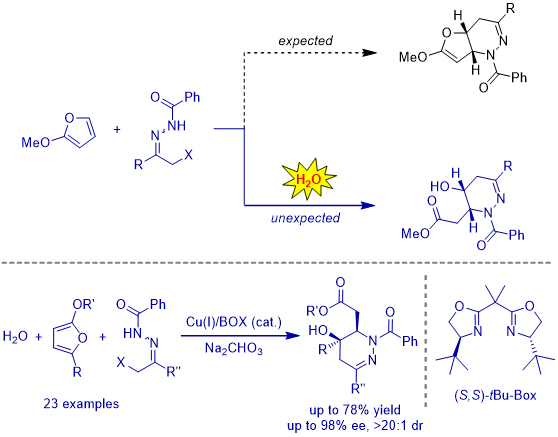
During the investigation of the behavior of furans in azoalkene-involved IEDDA reaction, we observed a novel Cu(I)-catalyzed asymmetric multicomponent cascade IEDDA/nucleophilic addition/ring-opening reaction. Replace indole with 2-methoxyfuran did not produce the anticipated bicyclic heterocycle. Instead, the cycloadduct was further attacked by water as nucleophile followed by tetrahydrofuran ring-open leading to highly unexpected but biologically attractive tetrahydropyridazine derivatives bearing g-hydroxyl ester moiety. Enantioenriched functionalized g-hydroxyl esters are versatile building blocks with two distinct functional groups for further synthetic manipulations. Notably, methodologies for those molecules containing g-hydroxyl ester moiety are almost unknown due to the propensity of g-hydroxyl ester to lactonize spontaneously.
 loading......
loading......